
Die Nachricht sorgte vor mehr als 20 Jahren für Schlagzeilen: Der Biotechnologe Craig Venter und das internationale Humangenomprojekt verkündeten nacheinander, erstmals die ganze Erbsubstanz eines Menschen sequenziert zu haben. Sequenzieren, das heißt: eine enorm lange Liste mit den vier Buchstaben A, T, G und C zu erstellen. Die Buchstaben stehen für Nukleinbasen, die in ihrer spezifischen Abfolge die Information der gesamten DNA darstellen. Quasi alles, was die Zellen eines menschlichen Körpers einmal produzieren müssen, ist auf diese Weise im Zellkern gespeichert – eingeschrieben mit den Basen Adenin, Thymin, Guanin und Cytosin.
Aber "vollständig entschlüsselt" bedeutet nicht für alle das Gleiche. Und durch den Versuch, den Code zu knacken – oder zumindest einmal im Ganzen aufzuschreiben –, stießen Forschende immer wieder auf neue Probleme. Nachdem es im Jahr 2000 hieß, das entschlüsselte Genom sei zu 97 oder 99 Prozent vollständig, wurde dieser Wert in den darauffolgenden Jahren immer wieder nach unten korrigiert. Mittlerweile stehen die Schätzungen zu den damaligen Arbeiten bei etwa 92 Prozent.
Die herausfordernden letzten Prozent
Die ausstehende Ergänzung wurde nun endlich vorgenommen, wie das renommierte Fachjournal "Science" aktuell berichtet, begleitet von etlichen zusätzlichen Studien. Bereits im vergangenen Jahr wurde eine entsprechende Forschungsarbeit auf einen Preprint-Server geladen, war also noch nicht von unabhängigen Fachleuten begutachtet. "Man sollte meinen, dass bei 92 Prozent des Genoms, die schon vor langer Zeit vervollständigt wurden, weitere acht Prozent nicht viel beitragen würden", sagt der beteiligte Forscher Erich D. Jarvis von den US-amerikanischen National Institutes of Health. "Aber durch diese fehlenden acht Prozent gewinnen wir jetzt ein völlig neues Verständnis davon, wie sich Zellen teilen, und können eine Reihe von Krankheiten untersuchen, die wir vorher nicht verstehen konnten."
Gerade diese fehlenden acht Prozent – mehr als 150 Millionen Basenpaare – waren nicht einfach zu untersuchen und hatten es wahrlich in sich. Denn dabei handelte es sich um Areale, in denen ein Buchstabenmuster etliche Male wiederholt wird. Das kann das richtige Ablesen des DNA-Strangs ziemlich durcheinanderbringen und für Fehler sorgen. Denn bei den üblichen Methoden wird der Strang in winzige Stücke zerlegt und wieder zusammengesetzt. Kommen nun immer wieder die gleichen Sequenzen oder muss eine ganze Reihe derselben Base gezählt werden, etwa viele "A"s hintereinander, erhöht sich die Fehlerquote bei solchen Technologien.
Mehr als Müll
Um das zu vermeiden, setzt die neue Studie eines Teams um Karen Miga von der kalifornischen Universität in Santa Cruz unter anderem auf zwei Methoden, die sich ergänzen. Während bei der einen vereinfacht gesagt lange DNA-Fäden am Stück abgelesen werden, scannt die zweite Technik die Abfolgen sicherheitshalber mehrmals ab. Die Arbeit mit diesen Werkzeugen wird in begleitenden Studien im Fachblatt "Nature Methods" vorgestellt. Manche Teile, die nur schwierig automatisch abzulesen sind, mussten aber auch "manuell", also von den Forschenden selbst, entwirrt werden, "so wie wenn man die Schnur eines Jo-Jos entwirrt", sagt Jarvis.
Wenn sich nun die Muster in diesen Bereichen andauernd wiederholen, könnte man sich fragen, ob sie für die Wissenschaft überhaupt interessante Informationen liefern. Und tatsächlich gingen viele Forschende zunächst davon aus, dass sich eine nähere Betrachtung kaum lohnt. Der Begriff "junk DNA", also "Müll-DNA", drängte sich zu Beginn der Sequenziertätigkeiten auf. Damit werden etwa Bereiche bezeichnet, deren Information nicht unmittelbar für die Herstellung eines bestimmten Proteins verantwortlich ist. Erst später kristallisierte sich heraus, dass diese nicht codierenden Teile des Erbguts ebenfalls eine wichtige Rolle spielen können: Einige von ihnen reglementieren indirekt, welche codierenden Gene gerade abgelesen und in Proteine übersetzt werden sollen.
Das Zentrum der Zellteilung
Im Zuge der neuen Studie zeigt sich die Forschungsgruppe überzeugt, dass der ergänzte Code wichtige Hinweise bezüglich bestimmter Krankheiten und sogar der menschlichen Evolution und Diversität liefern kann. Das hängt auch mit der Lage dieser sich wiederholenden Sequenzen zusammen: Sie befinden sich großteils in der Nähe des Zentromers, der die zwei Teile jedes Chromosoms mehr oder weniger mittig zusammenhält. Hier trennen sich duplizierte Chromosomen bei der Zellteilung. Die Stelle ist also beispielsweise bei der Entwicklung eines Zellhaufens zum Menschen besonders wichtig, sowie bei der Zellerneuerung. Und sie könnte bei Krankheiten wie Krebs, bei denen die Zellteilung außer Kontrolle gerät, ebenfalls essenziell sein.
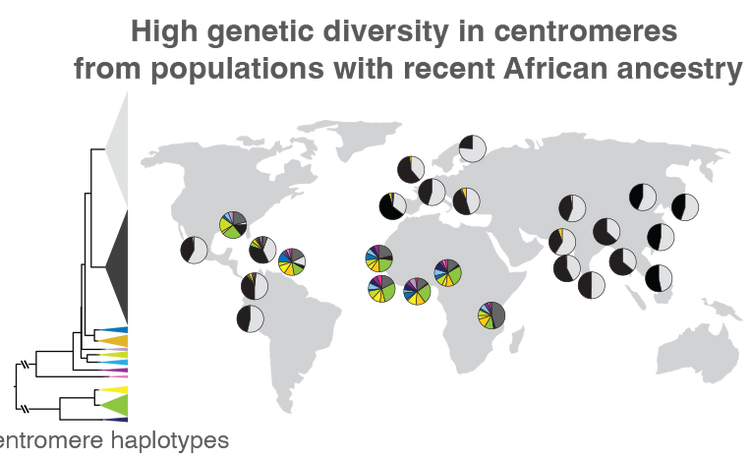
Aber nicht nur medizinische, sondern auch evolutionsbiologische Fragen dürften die nun entzifferten Areale beantworten, wie das an den Studien beteiligte T2T-Konsortium berichtet, das namentlich für das Abscannen von Chromosomen vom Anfangs- bis zum Endpunkt – also von Telomer zu Telomer – steht. In einer darauf aufbauenden Studie verglich eine der Forschungsgruppen die Zentromer-DNA von 1.600 Individuen auf der ganzen Welt. Sie stellte fest, dass Menschen mit rezenten afrikanischen Vorfahren in diesem Bereich eine viel größere genetische Variation aufweisen als etwa Menschen aus Europa und Asien ohne jüngere afrikanische Familiengeschichte. Künftige Studien könnten also an dieser Genomregion die menschliche Evolution und verschiedene Wanderungsbewegungen nachvollziehen.
Nur die halbe Geschichte
Ganz ohne Kritik kommt aber auch die angebliche Vollkommenheit dieses menschlichen Genoms mit dem Namen T2T-CHM13, das 3.055 Milliarden Basenpaare und fast 20.000 proteincodierende Gene umfasst, nicht aus. Ganz wie bei den Sequenzierungen vor 22 Jahren wurden nämlich auch jetzt nicht die 46 Chromosomen untersucht, die normalerweise in den Zellen eines Menschen stecken. Stattdessen wurden der Einfachheit halber quasi nur 23 Chromosomen analysiert – was der Menge entspricht, die eine Person von einem Elternteil vererbt bekommt. Sie besitzt also ein Paar der Chromosomen eins bis 22 plus Geschlechtschromosomen – bei Frauen in der Regel zwei X-Chromosomen, bei Männern ein X- und ein Y-Chromosom.
Bild nicht mehr verfügbar.
Die beiden Varianten eines Chromosoms enthalten vergleichbare Informationen. Aber genau genommen wurden bei dieser Studie eigentlich nur die Hälfte eines menschlichen Genoms abgelesen: Die Erbinformation entstammt nämlich einer Art menschlichem Tumor, der seine mütterliche DNA abgestoßen und die väterliche identisch verdoppelt hat. Dies vereinfachte allerdings die Sequenzierung.
Immer öfter werden bei Genomanalysen aber alle 46 Chromosomen menschlicher Zellen berücksichtigt – und das will die Forschungsgruppe nun ebenfalls nachholen, auch mit mehr als nur einem Genom, das als Referenz für die gesamte menschliche Vielfalt gelten soll. Ist das bewerkstelligt, werden vermutlich wieder einmal die Sektkorken knallen; immerhin werden diese Schritte erneut bemerkenswerte Meilensteine in der Geschichte der Genforschung darstellen. (Julia Sica, 31.3.2022)